A.7 Joins, or another way at looking at overlaps between Ranges
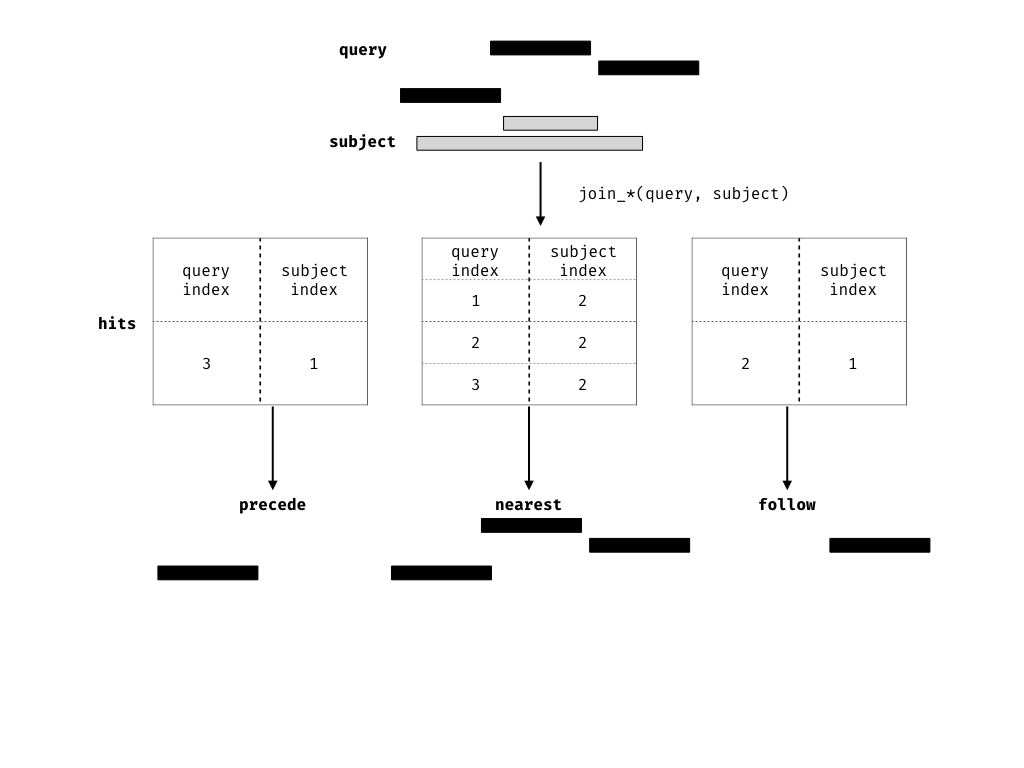
A join acts on two GRanges objects, a query and a subject.
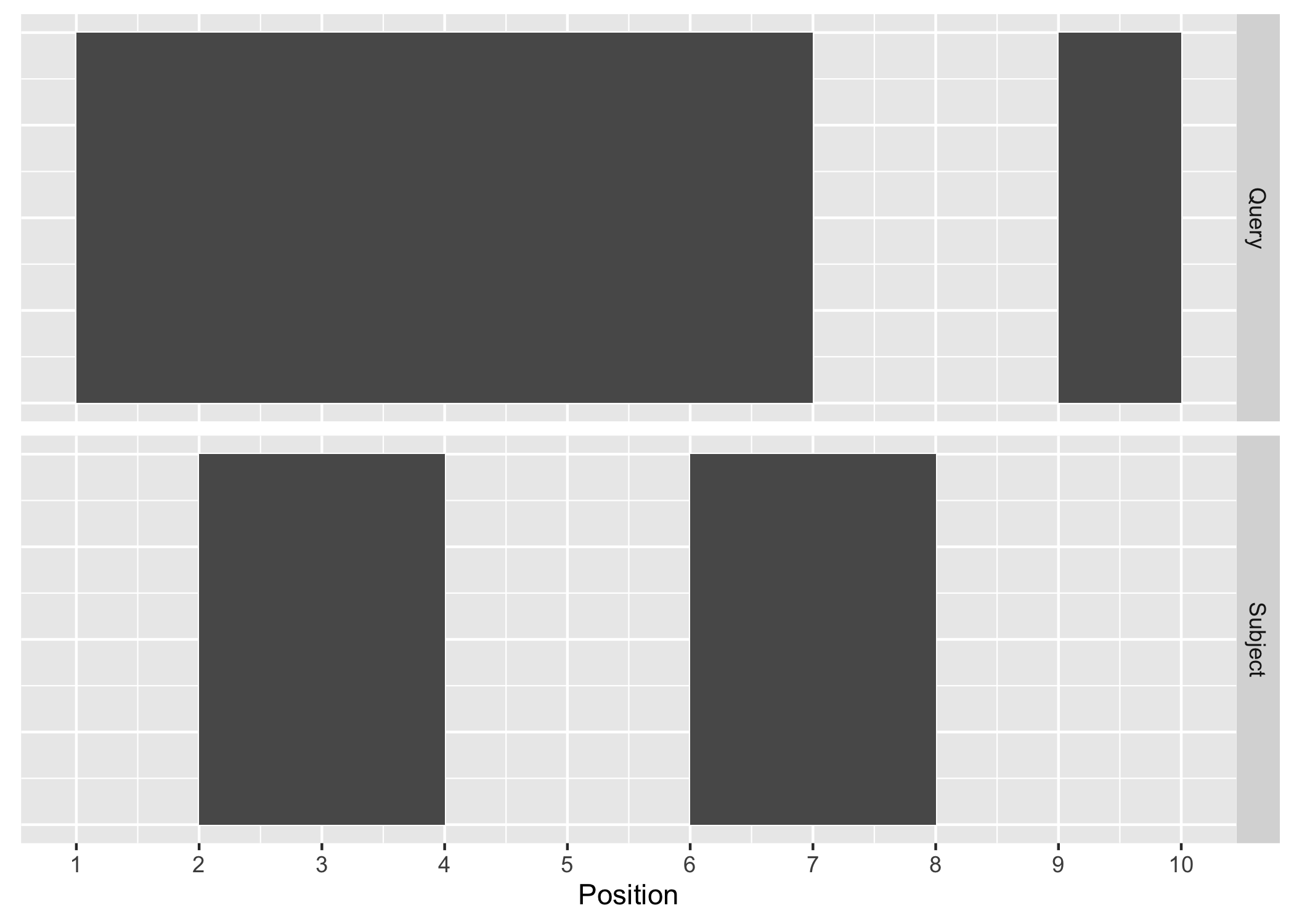
Figure A.1: Query and Subject Ranges
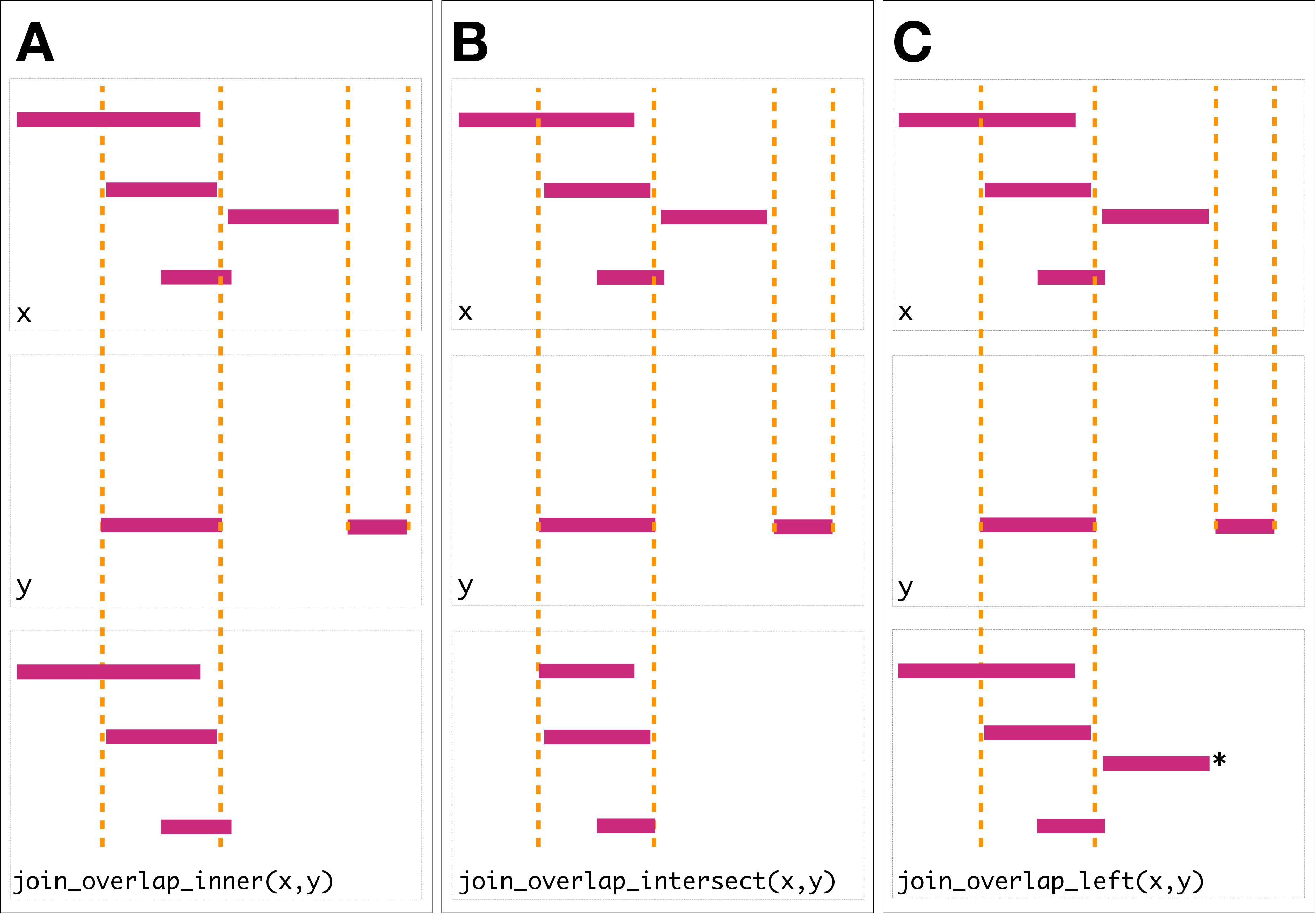
The join operator is relational in the sense that metadata from the query and subject ranges is retained in the joined range. All join operators in the plyranges DSL generate a set of hits based on overlap or proximity of ranges and use those hits to merge the two datasets in different ways. There are four supported matching algorithms: overlap, nearest, precede, and follow. We can further restrict the matching by whether the query is completely within the subject, and adding the directed suffix ensures that matching ranges have the same direction (strand).
The first function, join_overlap_intersect() will return a Ranges object
where the start, end, and width coordinates correspond to the amount of any
overlap between the left and right input Ranges. It also returns any
metadata in the subject range if the subject overlaps the query.
#> GRanges object with 2 ranges and 2 metadata columns:
#> seqnames ranges strand | key.a key.b
#> <Rle> <IRanges> <Rle> | <character> <character>
#> [1] chr1 2-4 + | a A
#> [2] chr1 6-7 + | a B
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths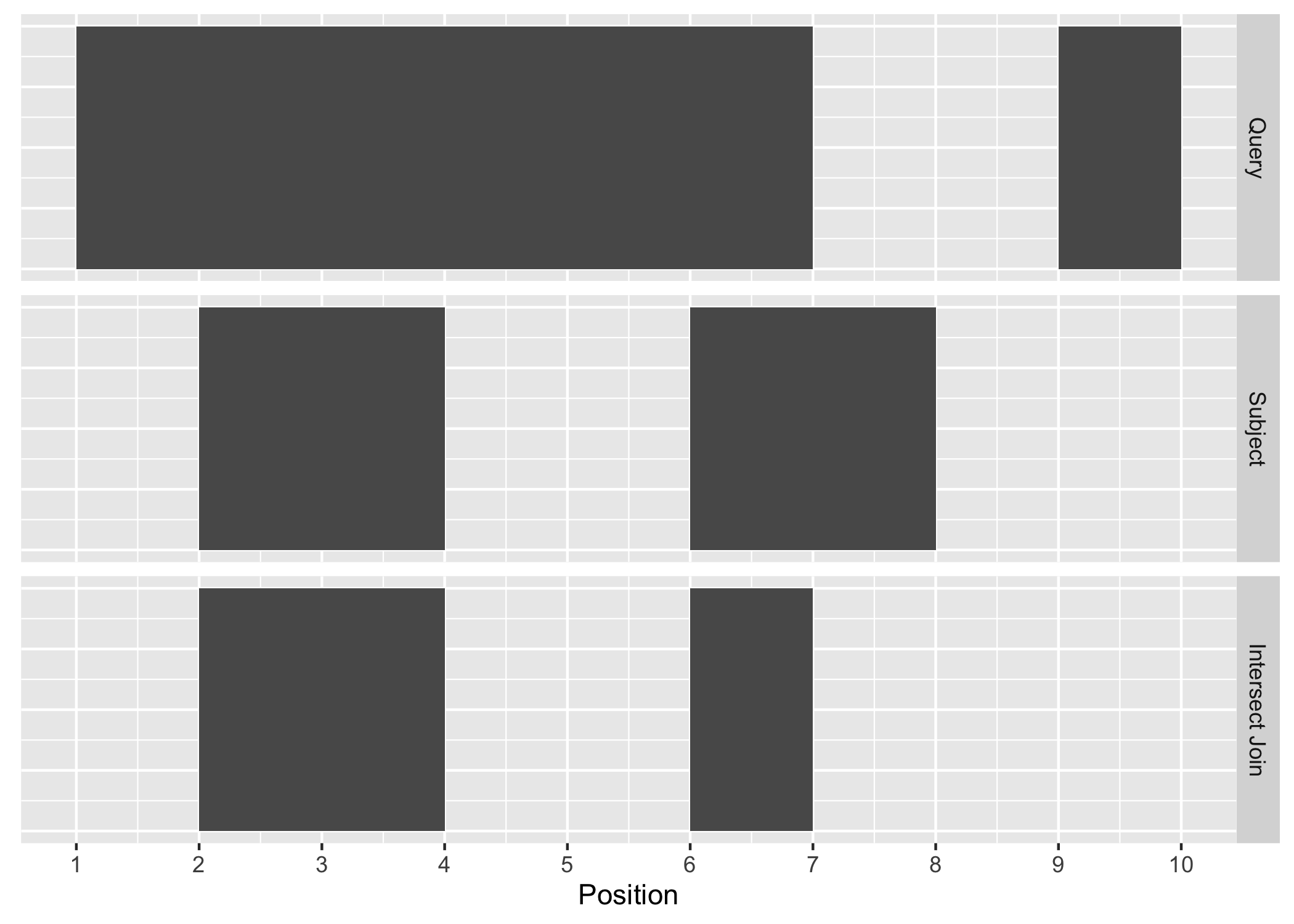
Figure A.2: Intersect Join
The join_overlap_inner() function will return the Ranges in the query that
overlap any Ranges in the subject. Like the join_overlap_intersect()
function metadata of the subject Range is returned if it overlaps the query.
#> GRanges object with 2 ranges and 2 metadata columns:
#> seqnames ranges strand | key.a key.b
#> <Rle> <IRanges> <Rle> | <character> <character>
#> [1] chr1 1-7 + | a A
#> [2] chr1 1-7 + | a B
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths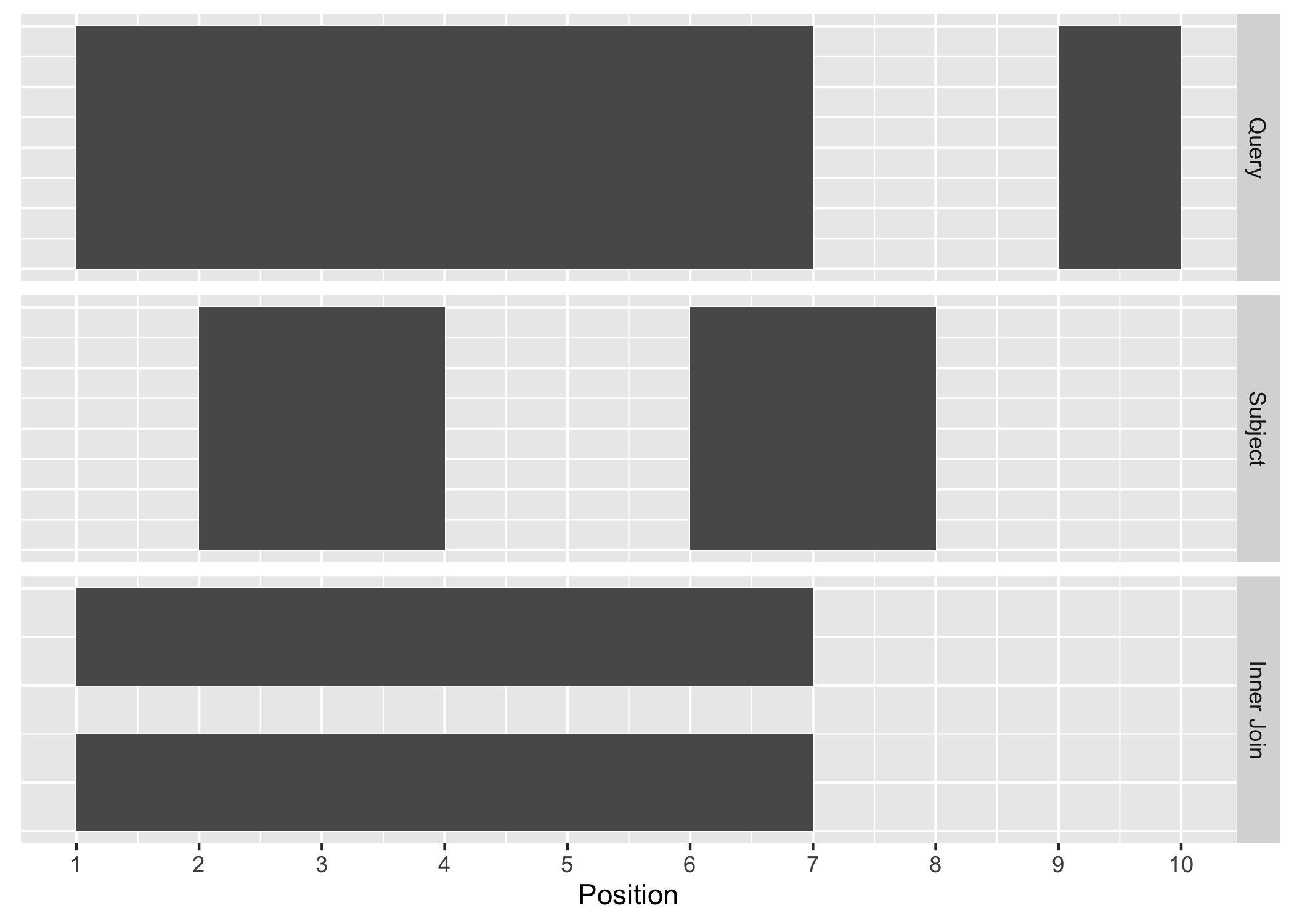
Figure A.3: Inner Join
We also provide a convenience method called find_overlaps that computes the
same result as join_overlap_inner().
#> GRanges object with 2 ranges and 2 metadata columns:
#> seqnames ranges strand | key.a key.b
#> <Rle> <IRanges> <Rle> | <character> <character>
#> [1] chr1 1-7 + | a A
#> [2] chr1 1-7 + | a B
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsThe join_overlap_left() method will perform an outer left join.
First any overlaps that are found will be returned similar to
join_overlap_inner(). Then any non-overlapping ranges will be returned, with
missing values on the metadata columns.
#> GRanges object with 3 ranges and 2 metadata columns:
#> seqnames ranges strand | key.a key.b
#> <Rle> <IRanges> <Rle> | <character> <character>
#> [1] chr1 1-7 + | a A
#> [2] chr1 1-7 + | a B
#> [3] chr1 9-10 - | b <NA>
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths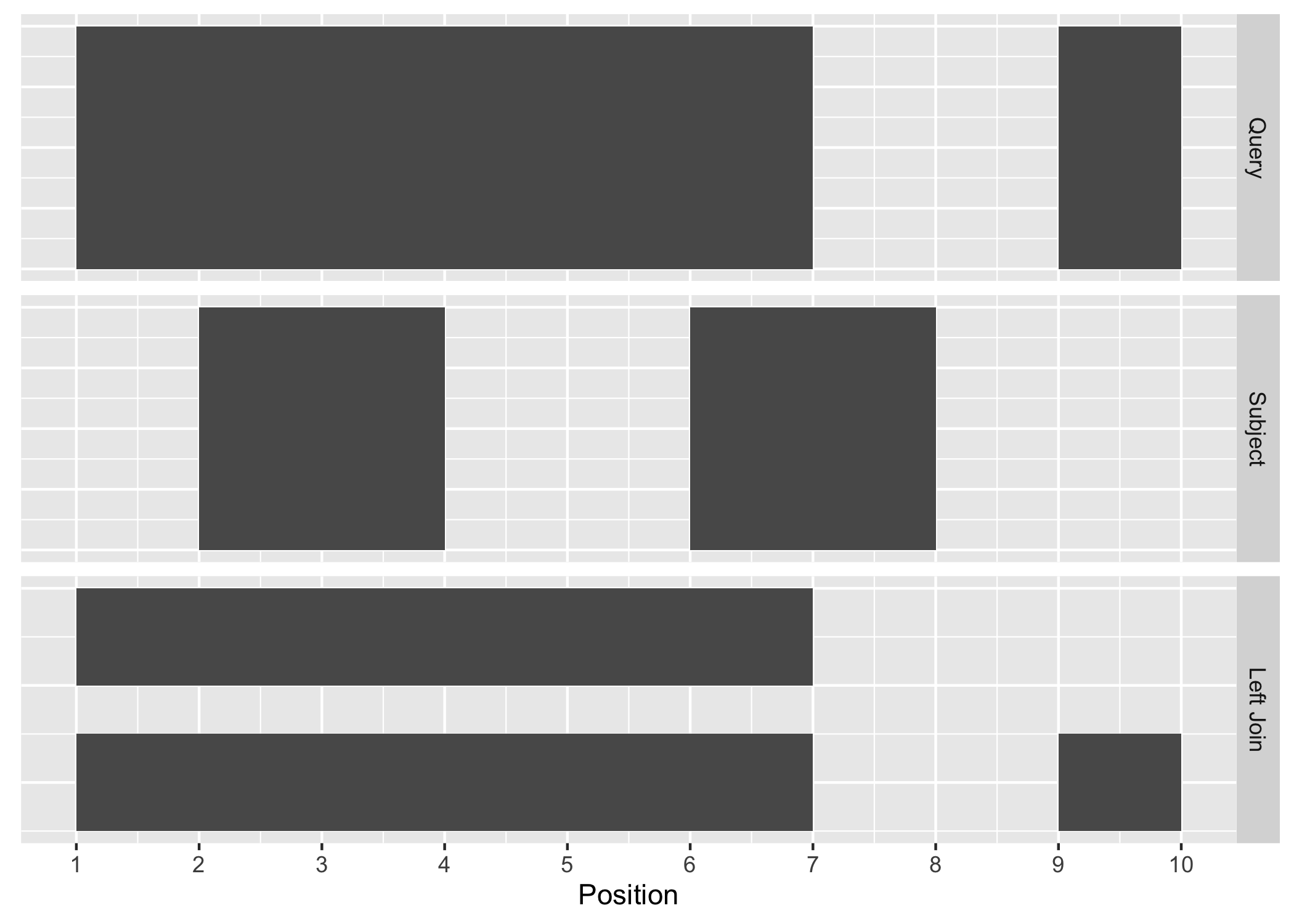
Figure A.4: Left Join
Compared with filter_by_overlaps() above, the overlap left join expands the
Ranges to give information about each interval on the query Ranges that
overlap those on the subject Ranges as well as the intervals on the left that
do not overlap any range on the right.
A.7.0.1 Finding your neighbours
We also provide methods for finding nearest, preceding or following Ranges. Conceptually this is identical to our approach for finding overlaps, except the semantics of the join are different.
#> IRanges object with 4 ranges and 1 metadata column:
#> start end width | gc
#> <integer> <integer> <integer> | <numeric>
#> [1] 5 9 5 | 0.780359
#> [2] 10 14 5 | 0.780359
#> [3] 15 19 5 | 0.780359
#> [4] 20 24 5 | 0.780359#> IRanges object with 4 ranges and 1 metadata column:
#> start end width | gc
#> <integer> <integer> <integer> | <numeric>
#> [1] 5 9 5 | 0.777584
#> [2] 10 14 5 | 0.603324
#> [3] 15 19 5 | 0.780359
#> [4] 20 24 5 | 0.780359#> IRanges object with 0 ranges and 1 metadata column:
#> start end width | gc
#> <integer> <integer> <integer> | <numeric>#> IRanges object with 5 ranges and 1 metadata column:
#> start end width | gc
#> <integer> <integer> <integer> | <numeric>
#> [1] 2 4 3 | 0.777584
#> [2] 3 6 4 | 0.827303
#> [3] 4 8 5 | 0.603324
#> [4] 5 10 6 | 0.491232
#> [5] 6 12 7 | 0.780359A.7.0.2 Example: dealing with multi-mapping
This example is taken from the Bioconductor support site.
We have two Ranges objects. The first contains single nucleotide positions corresponding to an intensity measurement such as a ChIP-seq experiment, while the other contains coordinates for two genes of interest.
We want to identify which positions in the intensities Ranges overlap the
genes, where each row corresponds to a position that overlaps a single gene.
First we create the two Ranges objects
#> GRanges object with 6 ranges and 0 metadata columns:
#> seqnames ranges strand
#> <Rle> <IRanges> <Rle>
#> [1] VI 3320 *
#> [2] VI 3321 *
#> [3] VI 3330 *
#> [4] VI 3331 *
#> [5] VI 3341 *
#> [6] VI 3342 *
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths#> GRanges object with 2 ranges and 1 metadata column:
#> seqnames ranges strand | gene_id
#> <Rle> <IRanges> <Rle> | <character>
#> [1] VI 3322-3846 * | YFL064C
#> [2] VI 3030-3338 * | YFL065C
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsNow to find where the positions overlap each gene, we can perform an overlap join. This will automatically carry over the gene_id information as well as their coordinates (we can drop those by only selecting the gene_id).
#> GRanges object with 8 ranges and 1 metadata column:
#> seqnames ranges strand | gene_id
#> <Rle> <IRanges> <Rle> | <character>
#> [1] VI 3320 * | YFL065C
#> [2] VI 3321 * | YFL065C
#> [3] VI 3330 * | YFL065C
#> [4] VI 3330 * | YFL064C
#> [5] VI 3331 * | YFL065C
#> [6] VI 3331 * | YFL064C
#> [7] VI 3341 * | YFL064C
#> [8] VI 3342 * | YFL064C
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsSeveral positions match to both genes. We can count them using summarise and
grouping by the start position:
#> DataFrame with 6 rows and 2 columns
#> start n
#> <integer> <integer>
#> 1 3320 1
#> 2 3321 1
#> 3 3330 2
#> 4 3331 2
#> 5 3341 1
#> 6 3342 1